


-

新闻和活动

蛋白类生物标记物在生物医药研发中的应用及生物分析策略
-

发布日期:2022-06-15
-

浏览量:0
-

发布者:
“生物标志物”一词最早出现在1947年关于胎球蛋白A检测方法的论文中[1],是一种客观测量并评价正常生物过程、病理过程或对药物干预反应的指示物,也是生物体受到损害时的重要预警指标。它涉及细胞分子结构和功能变化,生化代谢过程变化,生理活动异常表现以及个体、群体或整个生态系统的异常变化等[2]。小分子、蛋白类、细胞、组织学、放射成像或生理学特征等都是生物标志物的类型。比如:传统的生物标志物包括血压的可测变化、运动后血液中乳酸的浓度水平、糖尿病患者的血糖指标等。细胞中DNA、RNA、代谢产物或蛋白质含量水平在分子层面的具体变化等均可称为生物标志物。
根据2016年发布的BEST(Biomarkers, Endpoints, and other Tools)[3],生物标志物可分为:安全生物标志物、诊断生物标志物、易感性\风险生物标志物预后生物标志物、反应生物标志物、预测性生物标志物、监测生物标志物。比如:血清肌酐可用作安全生物标志物,用于评估患者使用影响肾功能的药物来监测肾毒性;血糖或血红蛋白A1c(HbA1c)可用作诊断生物标志物,以识别2型糖尿病(DM)患者。
生物标志物在药物开发的整个周期(药物发现、临床前、临床到上市)起着重要作用。在药物发现阶段:用于药物靶点选择、明确作用机制;在临床前阶段:可用于剂量选择、临床前安全性评估、明确作用机制;在临床阶段:可作为诊断分层、患者选择、剂量选择、药代动力学、安全性评估、疗效评估等;在上市阶段,可用于陪伴诊断试剂盒、临床安全性监测、临床有效性监测、药品质量控制等作用[4]。
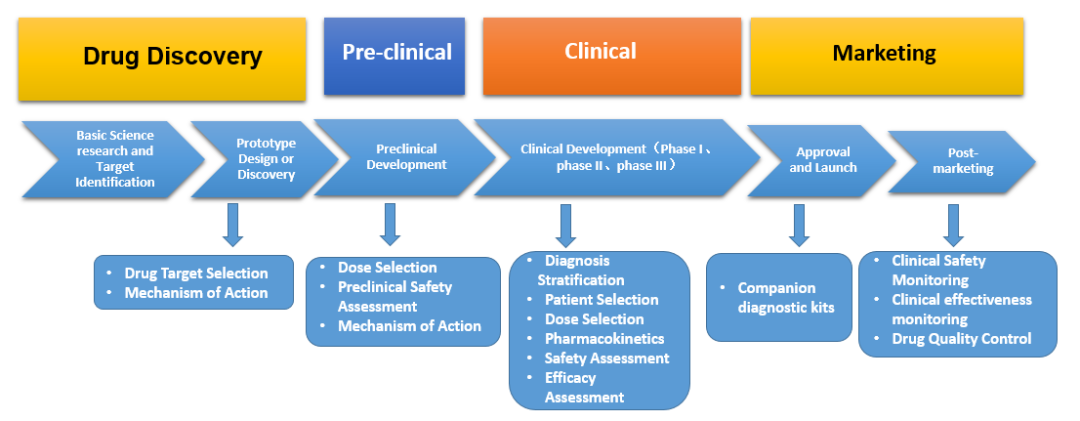
图1. 生物标志物在药物开发整个周期中的作用
2004年美国食品药品监督管理局(FDA)发布《创新/停滞:新医疗产品的关键路径上的挑战与机遇》白皮书提出生物标志物是提升新药研究开发能力的关键因素[5]。在新药研究开发过程中,生物标志物可有效提高研究决策,包括药物剂量、治疗时间、实验设计、风险/效益比、临床转化、临床受试人群的精准选择等方面,其指导治疗性候选化合物早期临床开发已被视为降低失败风险的一种有效手段。生物技术工业组织(Bio)公布的一份报告显示,具有生物标志物指导开发的新药获批成功率是没有生物标志物的3倍。
案例:在Nature子刊的一篇综述中[6]总结了临床试验失败的几个主要原因:错误的靶点、错误的分子、错误的结果和错误的病人。以上这些问题可以通过生物标志物引导的临床试验设计来有效解决,从而提高临床试验成功率。比如预测患者分层的生物标志物不仅有助于识别对药物有高反应的患者,还可以排除对药物副作用敏感的患者。
个性化治疗成功案例之一是基因泰克公司的乳腺癌治疗单抗赫赛[7]。Her2是人表皮生长因子受体2,发现于20世纪80年代,是肿瘤靶向治疗的重要靶点。赫赛汀曲妥珠单抗是一种抗Her2的人源单克隆抗体,可阻断Her2,破坏下游通道,抑制肿瘤细胞的生长。
众所周知,Her2在大约20%~30%的乳腺癌患者中过度表达。基因泰克首先开发了赫赛汀的药物。他们使用靶向Her2作为其预测生物标志物,在临床试验期间筛选入组患者。选取Her2表达过高的乳腺癌患者进行临床试验。赫赛汀的临床效率通过仅200名患者的III期临床研究证明,随即获得美国食品药品监督管理局(FDA)的加速审批获准上市,如果基因泰克当年没有采用Her2作为生物标记物来筛选潜在的有效病人,由于只有不到30%的乳腺癌病人有Her2过度表达,只有几百人的临床III期试验结果很可能会失败。生物标记物在药物研发中的作用可见一斑。
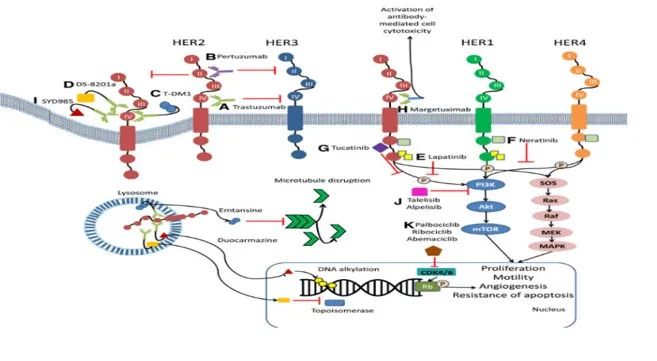
CA CANCER J CLIN 2020:70:355-374
图2. 曲妥珠单抗(赫赛汀,trastuzumab)作用机制
生物标记物涉及小分子、蛋白类、细胞、基因分子、组织切片等。通常可以用不同检测平台对不同种类的生物标记物进行测定:如用液质联用(LC-MS/MS)检测小分子类生物标记物(氢化可的松、雌激素、精氨酸);用配体结合方法(ligand binding assay)测定蛋白类生物标记物(炎症细胞因子白介素IL-6等);用流式细胞术(FACS)测定免疫细胞亚群的变化、受体占有率;免疫组织化学IHC用于组织染色(黑色素瘤切片,肿瘤活检标本肿瘤活检标本);q-PCR、dd-PCR用于基因组学生物标志物如(急性缺血性脑卒中:microRNA-27a-3p)等。
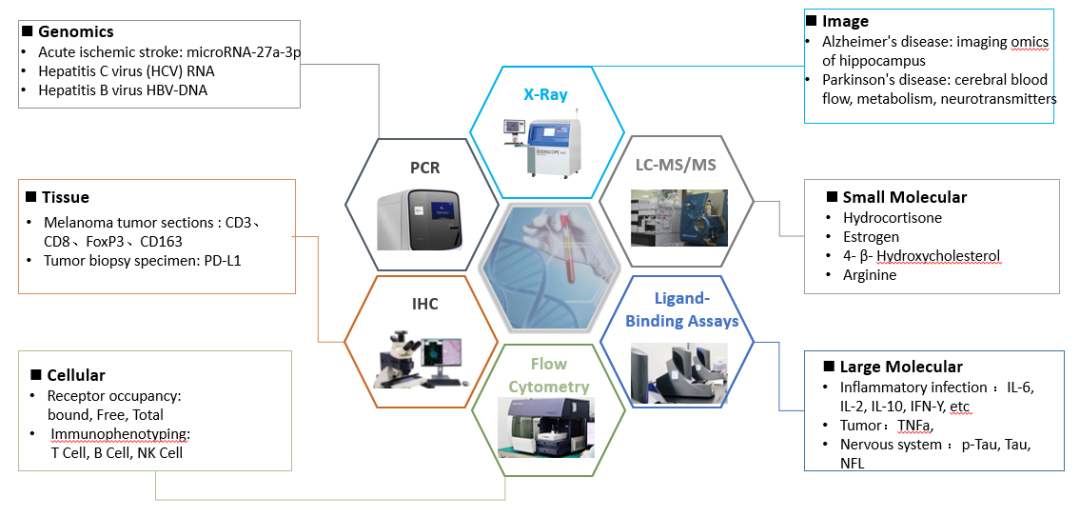
图3. 不同类生物标记物的检测平台
蛋白类生物标记物,通常使用配体结合方法(ligand binding assay)进行测定,常规的免疫检测分析平台如:酶标仪、电化学发光反应器MSD、Bio-Plex悬液芯片多重检测平台、Simoa全自动单分子免疫检测仪、Ella全自动微流控免疫学检测系统、SMCxPRO™单分子免疫检测平台等进行检测。
酶联免疫分析(ELISA)是很早被应用于体液样本临床蛋白标志物检测的技术,使用简单方便,在实验室应用广泛,已经使用了几十年,因而也被当成“金标准”来使用。酶联免疫分析类型包括:直接法(Direct ELISA)、竞争法(Competitive ELISA)、间接法( Indirect ELISA ) 和夹心法(Sandwich ELISA),但是ELISA灵敏度相对有限,线性范围窄。
电化学发光反应器(MSD)利用电化学发光原理,灵敏度高、背景低、线性范围广。同时通过每个小孔不同位点包被不同抗体来实现多重检测,一次可以检测10个多因子。
Luminex悬液芯片多重检测平台使用不同染色微球和流式细胞术原理,可实现同时检测100个生物标记物。

Simoa全自动单分子免疫检测仪利用数字PCR检测原理,将磁珠免疫复合物密封在236000个小孔中反应(每个小孔反应的体积是50飞升),采用数字化的计算方式,可以将检测灵敏度提高到传统ELISA的1000倍。
Ella全自动微流控免疫学检测系统将微流控、自动化检测、超敏荧光检测技术整合,彻底颠覆了传统的ELISA技术,可实现全程自动化、全程自动化、从样本前处理到结果1.5小时、灵敏度高、检测范围宽等优点。
其他如Singulex、Gyros、Elispot、Randox等多种免疫检测平台用于蛋白类生物标记物的测定。
生物标记物可以在药物开发的过程中被科学家和监督机构广泛用于各种目的。这些应用可以从探索性的假设生成,内部决策,到病人筛选或关键的临床决策。考虑其复杂性,法规部门并没有统一的生物标志物方法验证指南。
中国药典和欧洲药品管理局(EMA)等法规并没有明确针对生物标记物的内容[8-9]。FDA在2013年的《生物分析验证法规指南》中提到[10],对于生物标记物可以使用“fit-for-purpose” 的生物分析策略。根据生物标记物应用的目的来确定其生物分析验证的程度和内容。如果生物标记物的数据将用于法规部门申报:比如用于安全性或有效性的关键决定或者用于支持药物给药剂量的指导,那么方法需要做全验证。对于用于支持早期药物研发(候选物选择、内部决策、概念验证等),申办方可以根据需要来选择合适的验证内容。工业界也发表了不少文章来讨论如何进行“fit-for-purpose”的方法验证,详细描述了如何进行生物标志物的方法确认或验证[11]。
药明康德生物分析部对于生物标记物方法学验证有专门的标准操作规程(SOP)以及最佳实践指南(Best Practice)来定义如何进行生物标记物方法确认或验证。通常根据生物标志物的使用目的,分为三个级别:
级别1:非GLP方法的建立,这样的方法用于探索性的假设生成,样本检测的结果不会用于监管机构的任何决策。
级别2:方法确认,这样的方法用于探索性目的,但需要评估方法性能的几个参数,样本检测的结果将由监管机构审核,但不用来做重要决定。
级别3:方法验证,这样的方法是按照生物分析的法规来验证所有的参数,样本检测的结果将由监管机构审核,如评估药物是否有效的次要终点。
以上三个级别的生物标志物的开发、确认\验证具体考察内容如下:
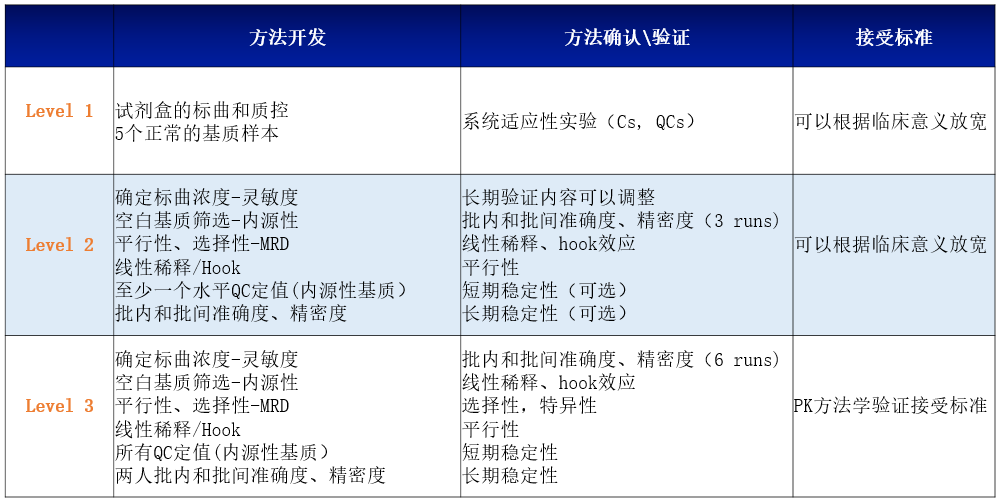
图5.三个级别的生物标志物的开发、确认\验证
对于级别1和级别2的生物标记物的方法学开发和确认内容,可以根据申办方的具体要求对考察内容进行一定的调整。
对于蛋白类生物标记物,通常可以用商品化的试剂盒或自主开发方法。商品化的试剂盒提供了快速的分析解决方案,操作方便、成本效益较低等,但由于不同供应商提供的试剂盒在方法开发和验证的内容以及关键试剂的性能把控方面差别很大,可能不足以支持临床药物研发的生物标记物的分析检测。而且作为商品化的试剂盒广泛应用于不同种属和不同基质,并非专门为临床应用而开发,因此尽管是商品化的试剂盒,在用于支持临床样本分析之前,仍需要做方法开发和验证。FDA和EMA的法规指南[9,10]也指出用于药物开发目的商品化试剂盒,仍需要进行验证以确保方法的可靠性。由于生物标记物的特殊性和复杂性,在方法开发和验证中会遇到下面不同的挑战:
对于蛋白质生物标志物的定量分析,通常是需要用蛋白质生物标志物的标准品来配制标曲。然而内源性的蛋白质生物标志物是在生物体内天然产生的,具有各种翻译后修饰的异构体,某种形式的异构体可以随疾病状态、遗传学、性别、年龄等变化[12]。而相反,不同供应商提供的不同标准品 (不同形式的异构体、重组蛋白、融合蛋白等)是纯化的、人工合成的,在蛋白序列、纯度、糖基化、折叠程度等方面,可能与内源性分析物不同,因此缺乏能够完整表征内源性分析物的生物标志物标准品,生物标记物分析是相对定量而不是绝对定量。在选择最能表征内源性生物标记物的标准品时,需要考虑以下几点:
内源分析物的理化性质 (蛋白质是否以单体形式存在,二聚体、三聚体等)
生物标志物的相关亚型,翻译后修饰、裂解、剪切等
外源性蛋白的表达系统 (如大肠杆菌,昆虫细胞或真核细胞表达系统)
外源性蛋白批次间可能的差异
不同厂家提供标准品的差异
因此最好尽量采用同一厂家、来源、批次的标准品,同时通过平行性实验考察内源性与替代性生物标记物差异,来确认外源性蛋白是否可以作为替代性生物标记物。
绝大部分的生物标记物都是内源性的,在生物体内可以检测到,因此标准品不能直接配制到空白的生物基质中,需要使用替代性的空白基质。通常可选下面的三种方式:
内源性低于LLOQ的空白基质,可以直接使用这些空白基质,这种方式的优点是基质差异小,处理简单。
使用去除内源性生物标记物的空白基质,比如:碳粉吸附、水解、亲和色谱等处理后的空白基质。优点是基质差异小,缺点是内源性生物标记物难彻底清除,处理方式繁琐、费用高。
使用异源基质(猴血清、幼儿血清)或者人工配置稀释液(牛血清蛋白)。人工配置稀释液的优点是具有更好的稳定性,适合长期使用。缺点是与内源性基质差异比较大,有时在基质效应方面不能模拟真实基质。
目前商品化的试剂盒,通常是人工配置稀释液(牛血清蛋白)做为空白基质,因此需要做平行性实验来考察替代性空白基质的可行性。
在方法验证和样本分析过程中,每个分析批都需要放置质控QC样本。质控QC样本需要模拟真实样本,最好使用内源性基质。由于很难找到高中低各个浓度的内源性基质,通常质控QC的配制有以下形式:
直接筛选合适浓度的内源性基质
用稀释液稀释后的内源性基质
内源性基质中直接添加标准品
上述质控QC样本至少需要通过3个批次的实验来确定浓度,用于方法学验证。当然对于级别1或级别2的方法学确认,也可在人工配置稀释液(牛血清蛋白)中添加标准品。一般试剂盒中会有这类质控QC,可以直接用于方法学确认,但是对于级别2的方法学确认,至少一个质控QC需要用内源性基质配制。
平行性实验在生物标记物方法学开发、验证中有非常重要的作用,是方法学考察中的一个非常重要的指标。正如前面提到,由于使用了替代性标准品和空白基质,所以需要考察其同内源性生物标记物的平行性。平行性实验通常在方法开发阶段就要进行考察,一般选取高内源性的多个不同来源的基质,进行1.5-2倍的序列稀释,并保证至少3-4个稀释后的浓度在线性范围之内,通过计算每个稀释浓度与最少稀释倍数(MRD)进行比较,80%以上需要满足回收率在100±20.0%和精密度要在20%之内。通过平行性实验可以确定MRD,评估分析的灵敏度,观察不同基质的选择性。
通常可以用健康受试者的基质来考察平行性,如果有些生物标记物在正常健康人中表达低,可以考虑使用病人受试者基质来考察平行性[13-14]。如果病人基质中的浓度也很低,最后可以考虑将标准品配制到空白基质中做序列稀释,作为平行性实验的参考。
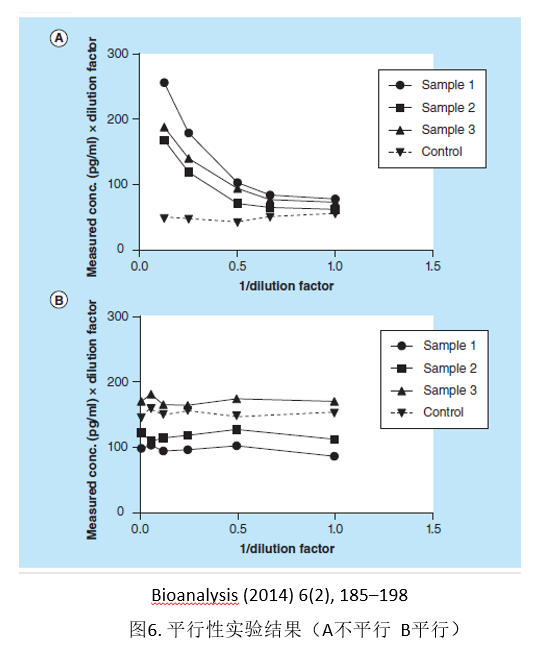
平行性的失败通常有两种情况:不同个体之间基质不平行和内源性基质同标曲不平行。不同个体之间基质不平行,可能是基质效应或者选择性问题引起。而内源性基质同标曲不平行,可能需要考虑更换标准品或者关键试剂。
在生物标记物分析方法验证中,考虑到外源性标准品只是替代性的生物标记物,因此选择性并不是必须要考察的指标。由于生物基质中往往含有内源性的生物标记物,因此选择性考察,需要采用加量法和减量法的方法来计算回收率。
加量法的计算公式:

减量法的计算公式:

美国药学科学家协会(AAPS)的一篇文献认为减量法相对加量法能够提供更加可靠的结果,并且建议添加外源性标准品的浓度至少是30%内源性基质浓度[15]。
用加量法和减量法考察选择性,要求至少80%以上的基质回收率在100±20.0%范围内。如果不能满足上述标准,可以通过基质加标后序列稀释的方法进行选择性考察。在至少6个批次的生物基质中加入已知量的标准品,进行1.5-2倍的序列稀释,并保证至少3-4个稀释后的浓度在线性范围之内,通过计算每个稀释浓度与最少稀释倍数(MRD)进行比较,80%以上需要满足回收率在100±20.0%以及精密度要在20%之内。
稳定性是考察检测的生物标记物在样本采集、运输、分析、储存过程中的稳定性。目前没有法规规定如何检测生物标记物的稳定性。常用有两种方法:内源性基质(单独或者混合)的稳定性和标准品配置到内源性基质中的稳定性[16]。但是标准品配置到内源性基质中的稳定性并不能代表真实样本的基质,不能真正模拟体内生物标记物,比如折叠和翻译后修饰的差异就可能导致稳定性的差异,例如标准品配制的IL-13基质中的稳定性只有5个月,而IL-13病人中的稳定性是15个月。
但是使用内源性基质来考察稳定性也会有问题。样本的稳定性是在不同时间分别测定后进行回收率比较,标准品或者关键试剂批号的改变可能会影响结果的测定。另外,疾病状态会影响蛋白酶调节的差异从而引起蛋白折叠的不同,可能也会影响内源性生物标记物的稳定性,例如TGF-B在正常人尿液中的冻融稳定性要高于病人尿液中的冻融稳定性。因此目前有文献报道可以使用计算机或者统计方法对内源性和配制的质控样本进行趋势和统计分析,来考察其稳定性。
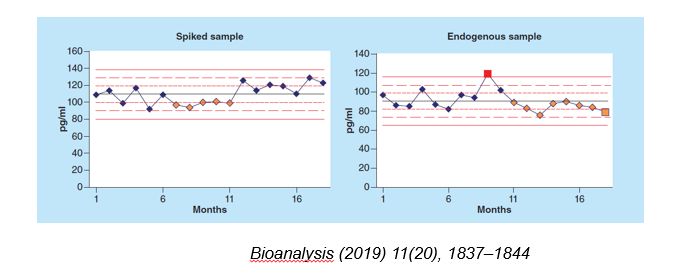
图7. 内源性和配制的质控样本趋势分析
随着技术发展,MSD、Luminex、Simoa、Ella等仪器平台都可以实现多重检测,允许在单个分析中同时测量多个分析物样本。多重检测好处:减少实验室分析时间、降低样品体积、节约成本等。但是在方法开发、验证以及样本分析过程中,由于其复杂性也带来了独特的挑战[17]。
定量范围和最小稀释倍数的确定:不同分析物的内源性水平不一致,所以样本的稀释倍数需要考虑其他分析物的稀释倍数。不同分析物平行性考察结果,也可能会产生不同的MRD。因此需要对不同化合物进行综合考虑,选取能够覆盖所有化合物的最小稀释倍数,但可能导致某些灵敏度低的分析物检测不到。
内源性基质QC的确定:由于不同分析物的内源性基质浓度不一样,有时很难找到一组合适的内源性基质QC用于所有需测定的分析物,因此需要配制不同组的内源性QC用于多个分析物测定,这样就降低了多重分析的优势和样本分析的方法适用性。
稳定性:样品稳定性受最不稳定的分析物的限制,如果一种分析物高度不稳定,建议将其从多因子测定的通道中移除。
不同分析物之间的交叉反应:由于多重检测具有多个捕获和检测抗体对,较单个分析物检测容易产生交叉反应,尤其是对具有相似的二级、三级结构或相似氨基酸序列的不同分析物,分析物的识别表位可能被其他分析物的抗体所识别,导致错误的信号,从而产生交叉反应。
串扰的影响:串扰是指来自单独分析物的信号可能会对另一种分析物产生影响。可通过配制不同浓度的某种分析物并将其他分析物保持在恒定的低水平浓度以及配制空白样品,通过信号比较来显示这种分析物对其他分析物背景信号的影响。如果确实存在串扰的问题,需要考虑更换试剂盒。
批间差异:关键性试剂的批间差异容易引起信号的变换。考虑到多重检测包含更多捕获和检测等关键试剂,更容易引起试剂盒批件的差异,建议购买足够数量的试剂盒或试剂进行验证和样品分析。如使用新试剂盒,要进行桥接批次的实验。
药明康德生物分析部在生物标记物检测方面已有17年的分析经验,完成了260个以上的生物标记物方法学确认或验证,并支持了120个以上新药的R&D开发及300个以上的临床实验。有非常丰富的生物标记物方法开发、验证以及分析经验。同时拥有先进而广泛的生物分析检测平台:涉及小分子、大分子蛋白类、细胞以及基因层面的生物标记物的检测。在蛋白类生物标记物测定方面,我们有各类先进的生物免疫检测平台(ELISA, MSD, Luminex, Randox,Gyros,、Ella, Elispot, Envision,Singulex等), 建立了各类细胞因子(白细胞介素、干扰素、肿瘤坏死因子、集落刺激因子、趋化因子、生长因子)以及多种疾病生物标记物的检测方法。如:银屑病(IL-23); 糖尿病(GLP、DPP-IV); 心血管类(CRP、IL-6、MCP-1); 神经类 (NFL、Tau); 肿瘤(EGFR/c-Met、CD19、HER2); 肾损伤(Cystatin C、BTK)。
2021年,药明康德生物分析部同 Quanterix建立了Simoa联合实验室,已完成9个检测丰度低、灵敏度要求高的生物标记物的方法学开发和验证(神经系统:NFL、 p-Tau-181、α-Synuclein、 Tau;炎症:IL-6、IFN-γ;癌症;TNFα、 GM-CSF;自体免疫疾病:IL-17A)。
[1]. Ultra centrifugal and electrophoretic studies on fetuin [J]. J Phys Colloid Chem, 1947, 51 (1):164.
[2]. Biomarker Applications in the Pharmaceutical Industry [M]// MATTES WB. The Path from Biomarker Discovery to Regulatory Qualification. Amsterdam: Elsevier INC. 2013, 12
[3]. 2020 BEST (Biomarkers, Endpoints, and other Tools) Resource, May 2018
[4]. 生物标志物在药品生命周期中的应用与展望, Acta Pharmaceutical Sinica 2021,56 (2): 456 -464
[5]. Innovation or stagnation: challenge and opportunity on the critical path to new medical products [EB/OL]. America: U.S. Food&Drug Administration, 2004 [2019-5-21]. http://www.fda.gov/Science Research/Special Topics/Critical Path Initiative/Critical Path Opportunities Reports/ucm077262.htm.
[6]. Reducing the risk of failure: biomarker-guided trial design [J]. Nat Rev Drug Discov, 2016, 15 (8): 517-518.
[7]. Epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors in breast cancer: current status and future development [J]. Front Biosci, 2005, 1(10): 2611-2617.
[8].中国药典2015年版.生物样品定量分析方法验证指导原则
[9]. Guideline on Bioanalytical Method Validation.European Medicines Agency, Committee for Medicinal Products for Human Use (CHMP), London, UK. (2011).
[10]. Draft Guidance for Industry. Bioanalytical Method Validation. US Department of Health and Human Services, US FDA (2013).www.fda.gov/downloads/drugs/guidancecompliance.
[11]. 2019 Critical Path Institute. Points to consider document: scientific and regulatory considerations for the analytical validation of assays used in the qualification of biomarkers in biological matrices.
[12]. Recommendations for Selection and Characterization of Protein Biomarker Assay Calibrator Material. The AAPS Journal, Vol. 19, No. 6, November 2017.
[13]. Parallelism: considerations for the development, validation and implementation of PK and biomarker ligand-binding assays. Bioanalysis (2014) 6(2), 185–198.
[14]. what is going on with my samples? A general approach to parallelism assessment and data interpretation for biomarker ligand-binding assays. Bioanalysis (2013) 5(16), 1941–1943.
[15]. Calculations for Adjusting Endogenous Biomarker Levels During Analytical Recovery Assessments for Ligand-Binding Assay Bioanalytical Method Validation. The AAPS Journal, Vol. 17, No. 4, July 2015.
[16]. New approaches for biomarker stability determination in regulated bioanalysis: trending, bridging and incurred samples. Bioanalysis (2019) 11(20), 1837–1844.
[17]. Development, validation, and implementation of a multiplex immunoassay for the simultaneous determination of five cytokines in human serum.Journal of Pharmaceutical and Biomedical Analysis 36 (2005) 1037–1044
关于生物分析部
药明康德测试事业部生物分析一站式服务平台旨在为全球客户提供全面的,专业的生物分析解决方案。药明康德生物分析部恪守全球最高质量监管标准,是符合GLP法规的检测机构,已多次通过国家药品监督管理局(NMPA)、美国食品药品管理局(FDA)、经济合作与发展组织(OECD)、欧洲药品管理局(EMA)、日本医药品医疗器械综合机构(PMDA)的全面核查。本部门拥有完善的大、小分子分析平台,为客户提供全方位的药物研发生物检测服务,拥有包括寡核苷酸、细胞基因治疗、双特异性抗体等新型药物形态的生物分析能力对于加速全球客户的新药研发进程、助力全球医药行业发展具有非常特殊意义。


